If there are 2 reactants:
A + B --> P
then v = k [A][B], and k is a second order rate constant, with units of conc-1 time-1 (M-1 sec-1)
Back to A--> P, ![]()
This says that the rate is equal to the disappearance of A, which is proportional to the concentration of A.
Now solve by intergration. First rearrange:
![]()
to become: ![]()
Now integrate from time t=0 to time t=t

![]()
or, ![]()
This is like radioactive decay-a reaction with no reverse reaction.
Half life is a constant. The time it takes to go from A(t) to 1/2 A(t + D) is a constant, D = ln2/k
This relationship can be shown if we substitute D = t into ![]()
ln(1/2Ao)/Ao = -k D
ln 1/2 = -kD
ln 2 = kD
D = (1/k)ln 2
D is called the half-life or t1/2
t1/2 = .693/k
In general enzyme reactions will be more complicated because some reverse steps must be considered.
Rates of Enzymatic Reactions (Historical note)
Enzyme kinetics:
Kinetic measurements of enzyme activity
- Routine characterization of enzyme preparations
- Elucidation of catalytic mechanisms
- Analysis of inhibitors
Many enzyme reactions can be described kinetically by this simple scheme:
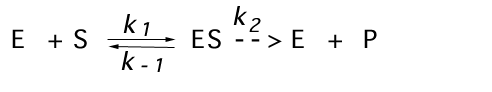
where E= enzyme, S= substrate, P= product, ES= noncovalent, enzyme-substrate complex (Michaelis complex)
In fact, many enzyme-catalyzed reactions are reversible ( i.e. k-2 can be significant), but at least initially, when [P]=0, the reverse rate will be zero.
What is the rate of this reaction? Since there is only one way to make P (ES--> P),
v=k2 [ES]
where k2 is a first order rate constant.
In general we do not know [ES]. What we do know is the total enzyme concentration, [E]T
[E]T = [E] + [ES]
and we do know [S] initially, which is approximately equal to [S] during early times of the reaction. (Another initial rate assumption)
We need some further assumptions to simplify the analysis.
In 1913 (see translation
of the original paper) Leonor Michaelis and Maud Menten
and Maud Menten  assumed that equilibrium of binding occurs:
assumed that equilibrium of binding occurs:
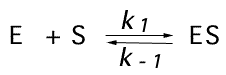 , so the dissociation constant Ks
is
, so the dissociation constant Ks
is
![]()
Because at equilibrium k1 [E][S] must equal k-1 [ES]
(The rate of the forward reaction must equal that of the reverse reaction.)
So, ![]()
Since,
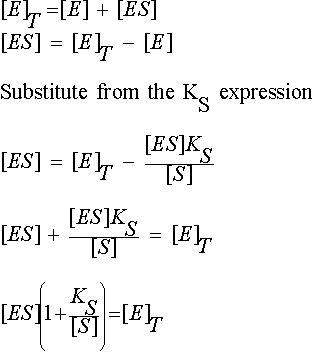
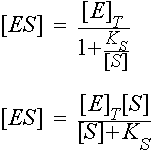
and since rate = k2[ES]
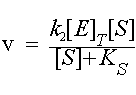
since the maximum value of [ES] is [E]T , the maximal rate is k2[E]T. This is called Vmax.
Vmax = k2[E]T
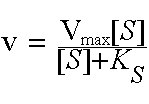
What if equilibrium of binding does not occur? (In general, if the k2 forward is faster than k-1 backward rate, then the binding is not in equilibrium. This is very common.)
Assume a steady state level of [ES], as was done by G.E. Briggs & J.B.S. Haldane (bio) (an interesting character) in 1925
This can be seen in Fig. 12-2. (See the Animation)
Write the rate equation for ES:
This reflects all of the things that can happen to ES:
It can be formed from E and S, so the first term
is ![]()
ES can dissociate back into E and S, so the next term is ![]()
Finally, ES can also release product P, so the last term is
![]()
In the steady state approximation, [ES] does not change, so:
![]()
So, ![]()
Since the Enzyme is either free enzyme, [E], or is substrate-bound enzyme, [ES]
Substitute for [E]: ![]()
then solve for [ES]:
![]()
![]()
![]()
![]()
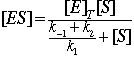
This leads to the rate, since ![]()
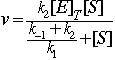
This gives the rate in terms of the total enzyme concentration, [E]T, the substrate concentration, [S], and the individual rate constants.
Note that this is of the form:
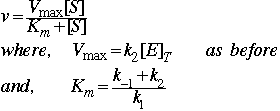
Remember the Michaelis-Menten assumption was that k-1 >> k2
In that case ![]()
This equation: 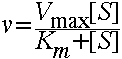 is called the Michaelis-Menten equation (although it is derived from the Briggs-
is called the Michaelis-Menten equation (although it is derived from the Briggs-
Haldane assumption). It says that the initial velocity of an enzyme reaction obeys M-M kinetics if it shows hyperbolic saturation in rate with increasing substrate concentration [S]. Note the similarity to O2 binding by Mb. Here Km is the substrate concentration at which v is equal to one-half of Vmax. Many enzymes obey M-M kinetics, even though there are additional intermediates and rate constants. In general, Km is a function of first and second order rate constants.
Fig. 12-3 Shows the hyperbolic dependence of rate on substrate. (See the Animation)
Analysis of Kinetic Data
We could use the Hill equation to get a straight-line plot of enzyme rate measurements, but the Lineweaver -Burk plot is a little simpler and is more commonly used. Typically, the rate (v) is measured at several different concentrations of substrate (S). By plotting these results, both the Km and the Vmax can be determined.
Lineweaver-Burk plot (double reciprocal) is derived from the Michaelis-Menten equation above.
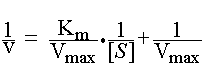
Here, ![]() is plotted versus
is plotted versus ![]() , which yields a straight line. The y-intercept is 1/Vmax. If the line is extrapolated to the x-axis, the x-intercept is -1/Km.
, which yields a straight line. The y-intercept is 1/Vmax. If the line is extrapolated to the x-axis, the x-intercept is -1/Km.
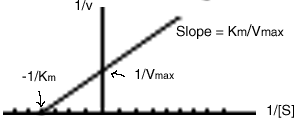
See the Animation of Fig. 12-4, the Lineweaver-Burk plot.
See Lesson 12-1 on the CD-ROM
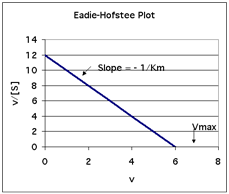
A second type of plot of kinetic data is called an Eadie-Hofstee plot.
Multiply the Michaelis-Menten equation to yield:
![]()
Now divide by [S] ![]()
Rearrange: ![]()
In the Eadie-Hofstee plot: v/[S] versus v.
The slope is -1/Km and the x-intercept is Vmax.
Back to 5310 Home
Last updated
Comments/questions: svik@mail.smu.edu
Copyright 2002, Steven B. Vik, Southern Methodist University